|
|
|
|

Retinite Pigmentosa: Introduzione
Con il termine retinite pigmentosa (RP) si indica ancora oggi
un gruppo di malattie ereditarie della retina che provocano una perdita
progressiva della visione notturna e del campo visivo periferico, e che sono
caratterizzate nella maggioranza dei casi dalla migrazione di pigmento nella
neuroretina, attenuazione dei vasi sanguigni retinici e pallore del disco
ottico. In molti casi vi è una perdita dell'acutezza visiva, che può
condurre all'ipovisione e progredire fino alla cecità.
La diagnosi deve essere sempre confermata da un elettroretinogramma
anormale od estinto.
Anche se il termine è incorretto
(queste malattie non sono "retiniti"), viene ormai usato universalmente, e la
tendenza è a mantenerlo.
I recenti risultati degli
studi di genetica molecolare sulla RP hanno dimostrato che esiste un comune
meccanismo patogenetico, la degenerazione primitiva dei fotorecettori, che
avviene sulla base di mutazioni di alcune delle proteine che costituiscono il
ciclo della visione.
Vengono differenziate le retiniti
pigmentose in cui la malattia retinica è unica manifestazione, dalle retiniti
pigmentose associate ad alterazioni di altri apparati, o sindromiche.
Esistono inoltre delle forme non genetiche di degenerazione
retinica molto simili alla RP che insorgono nei soggetti predisposti in seguito
ad alcune infezioni (sifilide, morbillo), all'introduzione di farmaci
retinotossici (clorochina, tioridazina, cloropromazina, indometacina,
tamoxifen), od a traumi oculari, le quali vengono denominate pseudo-retiniti
pigmentose.
Non sono da includere, invece, nel gruppo
della RP le malattie che pur avendo un meccanismo patogenetico analogo non
provocano la degenerazione dei fotorecettori e quindi la cecità, e non hanno
carattere progressivo, come l'emeralopia congenita stazionaria, e le distrofie
maculari ereditarie, che meritano una classificazione a parte.
Negli ultimi anni il progresso delle conoscenze in campo
genetico ha permesso di intravedere nuove possibilità di prevenzione e cura
delle malattie ereditarie, e quindi l'attività di studio si sta
indirizzando alla comprensione dei meccanismi genetici che provocano tali
degenerazioni retiniche.
Infatti l'identificazione del
gene/i responsabili della malattia rende possibile la effettuazione di programmi
di prevenzione attraverso l'identificazione dei portatori e costituisce la
premessa per una futura terapia genica, ossia per la correzione del materiale
genetico alterato.

 Ereditarietà
Ereditarietà
La Retinite Pigmentosa, secondo le statistiche internazionali, colpisce circa una persona su 4.000 sane, però la sua diffusione è ancor più nelle isole, nelle valli ed in tutte quelle comunità ove siano frequenti i matrimoni tra consanguinei. Molto spesso essa compare tra la pubertà e l'età matura, ma non sono assolutamente rari gli esempi di bambini colpiti nella prima infanzia.
La percezione che la retinite pigmentosa avesse delle base genetiche derivò, analogamente a quanto accadde per le altre malattie ereditarie, dalla osservazione della sua familiarità, ovvero dalla constatazione, invero psicologicamente impegnativa, che in molti casi la malattia si ripeteva fra i componenti di un ceppo familiare.
Ben presto si colse in questa ripetizione, non solo una più alta frequenza della malattia fra i familiari di soggetti affetti rispetto ai familiari di sani, oppure rispetto alla sua frequenza nella popolazione generale, ma si accertò anche che la malattia, quando si ripeteva, lo faceva seguendo delle leggi esprimibili in termini matematici. In definitiva si concluse che molti casi di retinite pigmentosa seguivano le leggi genetiche di Mendel.
Le forme genetiche di RP sono essenzialmente quattro: autosomica dominante, autosomica recessiva, legata al cromosoma X, e sporadica.
Per schematizzare questo reperto, si possono proporre i tre "modelli" di eredità che seguono per l’appunto le leggi di Mendel, nel modo seguente:
Nell’ambito delle famiglie colpite la malattia segue le seguenti regole:
a) può colpire maschi e femmine con pari frequenza;
b) non salta le generazioni .
Invero la eredità autosomica dominante prevede che ogni persona affetta abbia almeno un genitore affetto ed uno dei due nonni (per parte di quel genitore) a sua volta affetto. Questo schema però resta spesso teorico per diversi motivi: talvolta, soprattutto per le generazioni lontane, non veniva formalizzata la diagnosi, magari perché la malattia aveva una gravità clinica modesta, oppure un esordio tardivo; non si può neppure tralasciare l’evenienza che la malattia "cominci" in una persona che ha genitori e parenti sani per neomutazione di un gene.
Nel contesto di tutte le retiniti pigmentose la frequenza di quelle che si ereditano secondo il modello autosomico dominante varia da regione a regione: dal 10% circa ( in territorio svizzero) al 39% (nel Regno Unito); mediamente si propone un valore del 25% .
La manifestazione familiare di questo tipo di eredità è la seguente:
a) la malattia colpisce con pari frequenza entrambi i sessi ;
b) la malattia salta le generazioni, anzi non è infrequente l’evenienza che in una famiglia coinvolta compaia a memoria d’uomo un solo caso, ovvero che sia simulata una forma sporadica.
Questo comportamento familiare è riferibile al fatto che la persona affetta riceve il gene patologico da entrambi i genitori, i quali peraltro sono portatori sani. Dunque la nascita di un soggetto affetto dalla forma recessiva prevede che si siano incontrati due genitori portatori: questa evenienza è molto rara stante la bassa frequenza del gene, però è spesso resa sensibilmente più probabile dalla consanguineità dei due genitori oppure anche dalla loro comune provenienza da un piccolo centro.
Queste due constatazioni sui genitori di un soggetto affetto fanno propendere per la forma recessiva anche quando la persona in causa è l’unica affetta nella famiglia.
La proporzione delle forme recessive nel contesto di tutte le retiniti va dal 80% nel territorio svizzero al 15% nel Regno Unito: mediamente si stima attorno al 35% .
Secondo questo tipo di trasmissione ereditaria, risultano colpiti dalla malattia solo soggetti di sesso maschile, i quali però ereditano il gene patologico dalla madre che è portatrice sana; data una donna in tale condizione, il rischio di malattia per ogni figlio maschio è del 50% .
In una famiglia nella quale compaiano più casi della malattia, la percezione della eredità legata al sesso è in genere facile. Al solito se in un ceppo familiare compare un solo soggetto affetto di sesso maschile, è problematico stabilire se si tratti di eredità legata al sesso oppure autosomica recessiva o di un caso sporadico.
La proporzione dei casi di eredità legata al sesso varia da 1% (Svizzera e Russia) al 15% (Regno Unito); il valore che si propone mediamente è del 10% circa.
Le forme sporadiche ( circa il 30% di tutti i casi) prevedono dunque la presenza di un unico caso a memoria d’uomo in una famiglia. La sporadicità è solo una constatazione familiare, ma è molto difficile escludere la eredità recessiva oppure quella legata al sesso, se la persona affetta è di sesso maschile.
Il fatto che molti casi di retinite pigmentosa seguano uno degli schemi mendeliani di trasmissione ereditaria, dimostra che in quei casi la malattia è dovuta alla mutazione, ovvero ad una anomalia strutturale, di un gene; il gene mutato è la causa della malattia . Sono state scoperte numerose mutazioni di diversi geni, tutte capaci di determinare la retinite pigmentosa, tanto che, entro questa terminologia clinica, sono comprese molte forme geneticamente diverse.
Il campo nel quale le conoscenze sono ben lontane dall’essere esaurienti è quello della "patogenesi", ovvero dei meccanismi mediante i quali la mutazione di in gene determina l’insorgenza e la evoluzione della malattia. Dunque si conoscono molte (fin troppe) cause della retinite pigmentosa, ma non è noto come agiscano.
Si può , almeno in prima istanza, proporre questa sequenza: la mutazione di un gene determina una anomalia strutturale di una delle proteine che in qualche modo partecipa alla funzione visiva; da questa anomalia deriva la degenerazione delle cellule in cui la proteina è attiva e quindi la malattia.
Nei confronti della retinite pigmentosa ha un grande valore pratico la prevenzione , la quale si esercita mediante la consulenza genetica.
A sua volta quest’ultima consiste in una raccolta approfondita delle notizie familiari della persona affetta e nell’applicare ad esse le leggi genetiche già illustrate.
Spesso si rendono utili accertamenti clinici strumentali di competenza oculistica.
E’ utile dare una dimensione alla applicabilità delle tecniche di diagnosi molecolare del DNA, le quali tecniche peraltro sono troppo spesso enfatizzate e generano illusioni nelle persone coinvolte da questo problema.
Per riconoscere la presenza di una mutazione che può essere causa di retinite pigmentosa in un soggetto clinicamente sano (per esempio per trarne una definizione di rischio per i suoi discendenti), bisogna sapere quale mutazione cercare fra tutte le numerose possibili.
La strategia conoscitiva ed applicativa che dovrebbe essere seguita sarebbe quella di individuare la mutazione sicuramente presente nei soggetti affetti da una forma ereditaria, per poterla poi cercare nei membri sani delle loro famiglie.
L’approfondimento e la estensione delle conoscenze del DNA quali derivano dal progetto genoma, porteranno certamente a risultati significativi in questo campo.
Epidemiologia
La RP, nel suo complesso di varianti cliniche non sindromiche e
sindromiche, ha una prevalenza abbastanza variabile nelle varie popolazioni
studiate: negli USA, è di circa 1:3500-1:4000, con variazioni significative nei
vari Stati, in Svizzera 1:7000, in Cina 1:4016, in Norvegia 1:4440 in
Israele 1:4500.
Frequenza globale mondiale (varianti non sindromiche e
sindromiche): 1 caso ogni 3.000-5.000 abitanti (circa 1 milione e mezzo di casi
nel mondo).
Classificazione
Nonostante la similarità del quadro clinico delle varie forme di
RP, una classificazione unitaria soddisfacente non è mai stata trovata, a
dimostrazione delle notevoli differenze che in realtà esistono tra forme solo
apparentemente simili. Va ricordato che la RP è un fenotipo molto diffuso che
rappresenta il punto di arrivo comune di molte e diverse malattie retiniche.
Innanzitutto bisogna differenziare la RP primaria, in cui esiste solo
l'interessamento oculare, dalla RP che si ritrova associata a malattie
extraoculari.
La RP primaria, da un punto di vista fisiopatologico,
viene suddivisa classicamente in due gruppi principali: le forme in cui la
perdita di funzione dei bastoncelli precede quella dei coni ("rod-cone"), e le
forme in cui la perdita di funzione dei coni precede qella dei bastoncelli
("cone-rod").
Preliminarmente è importante tuttavia classificare
geneticamente le varie forme di RP, sia per fornire una prognosi di
massima al paziente definendo in prima approssimazione la gravità della forma di
retinopatia, sia per una valutazione delle probabilità di trasmissione ai
discendenti, sia per poter suddividere la malattia in sottogruppi relativamente
omogenei, che presumibilmente potrebbero avere un meccanismo patogenetico
comune, quadri fisiopatologici caratteristici, e la cui categorizzazione
faciliterebbe le indagini genetiche.
La RP autosomica dominante teoricamente
rappresenta il gruppo delle forme meno gravi. Da un punto di vista clinico viene
suddivisa in due tipi: tipo diffuso (D) con esordio precoce, e tipo regionale ®
con esordio ad età variabile, ma generalmente più tardivo. Tale
classificazione corrisponde entro certi limiti ai 2 gruppi ad esordio
precoce (tipo I) ed esordio tardivo (tipo II) dell'emeralopia di Massof e
Finkelstein . Nel tipo D la perdita di funzione fotorecettoriale riguarda
diffusamente tutto il fondo oculare e sono prevalentemente interessati i
bastoncelli, mentre nel tipo R la perdita di funzione riguarda sia i
bastoncelli che i coni, ma in forma localizzata, in aree determinate
circoscritte del fondo. Alcuni AA. hanno proposto l'introduzione anche della
forma a settore come terzo sottotipo.
La RP legata al cromosoma X è tipicamente la forma più severa in
termini di precocità di esordio, penetranza completa, progressione relativamente
rapida, alta incidenza di miopia e di cataratta. Più frequentemente in questo
tipo rispetto agli altri tipi di trasmissione l'acutezza visiva è ridotta a meno
di 5/10 tra l'età di 20 e 39 anni, l'esordio della emeralopia è inferiore ai 20
anni, esiste una miopia superiore alle 2 diottrie, ed un elettroretinogramma
praticamente estinto.
In un piccolo numero di famiglie con RPX tuttavia la
malattia è relativamente mite nei pazienti maschi, probabilmente per ragioni di
eterogeneità allelica o genetica. In alcuni casi, questi pazienti presentano una
buona acutezza visiva ed un campo visivo conservato anche dopo i trent'
anni, situazione assai rara in generale in questa forma di RP. Alcuni di questi
casi sono stati classificati come RP3 mediante analisi di linkage (vedi oltre).
In un certo numero di famiglie (41%) con RPX una percentuale significativa di portatrici (30%) della malattia mostra all'esame oftalmoscopico un caratteristico riflesso retinico nella regione para-maculare, denominato riflesso tapeto-retinico. Il fondamento anatomo-patologico di questo riflesso non è noto.
Delle varie forme genetiche di Retinite Pigmentosa
(non-sindromiche e sindromiche) sono stati localizzati oltre trenta loci
distinti, ed almeno nove geni responsabili.
La
maggioranza di questi geni codifica per proteine implicate nel ciclo della
visione, e la mutazione genica più frequente, sia in termini di numero di
mutazioni identificate (oltre 70), che di numero di pazienti colpiti (circa il
10%), riguarda il gene della rodopsina (il pigmento visivo dei bastoncelli)
localizzato sul cromosoma 3 (3q21-q24), ed associato pressochè esclusivamente
alla forma autosomica dominante (RP4).
In questa forma
di RP sono state identificate mutazioni a carico di un altro gene, la
periferina, localizzato sul cromosoma 6 (6p21.1-cen), sia in forma isolata che
digenica, in associazione con ROM1. Le mutazioni della periferina sono tuttavia
più frequentemente osservate nella cd. distrofia retinica maculare tipo
"pattern" od "a farfalla". Altri loci genici della forma autosomica dominante
sono stati identificati sui cromosomi 7, 8, 17 e 19.
Per la RP autosomica recessiva (RPAR) sono state identificate solo
quattro mutazioni, che insieme, sono presenti in meno del 10% dei pazienti
affetti, riguardanti il gene della rodopsina, della subunità b della
fosfodiesterasi GMPc (PDEB), situato nella banda cromosomica 4p16, che
rappresenta il sito più frequente di mutazione della RPAR (4% circa), il gene
dei canali GMPc dei bastoncelli, ed il gene della subunità a della
fosfodiesterasi GMPc (PDEA), situato sul cromosoma 5q31.2-q34. Due altri loci
responsabili della RPAR sono stati identificati mediante analisi di linkage in
famiglie con matrimoni tra consanguinei, la RP12 sul cromosoma 1q31-q32.1, e la
RP14 sul cromosoma 6p21.3 (distinto dalla periferina) e la RP16 sul cromosoma
14.
Nella forma di RP legata al cromosoma X (RP-X) gli
studi di linkage effettuati dimostrarono inizialmente l'esistenza di due diversi
loci codificati a seconda della posizione sul cromosoma X come RP2 (Xp11.3) ed
RP3 (Xp21.1), ma successivamente sono stati evidenziati altri due loci (RP6 e
RP15) nella regione distale di Xp.Del tutto recentemente nella RP3 è stato
possibile effettuare la clonazione posizionale di un gene, l'RPGR (retinitis
pigmentosa GTPase regulator), il cui prodotto proteico è rappresentato da una
proteina di 90 kD con funzioni regolatrici sulle piccole proteine leganti
GTP.
Classificazione genetica della retinite
pigmentosa primaria
________________________________________________________________________________
Tipo
Localizzazione
simbolo
&
Gene
identificato
Rif
cromosomica
n OMIM
________________________________________________________________________________
Autosomica Dominante (ad)
RP1 cromosoma
8q11-q21
180100
RP4 cromosoma
3q21-q24 RHO;
180380 rodopsina
RP5
(eliminata)
180102
RP7 cromosoma
6p21.1-cen RDS;
179605 periferina
RP8
?
RP9 cromosoma
7p15.1-p13
180104
RP10 cromosoma
7q
180105
RP11 cromosoma
19q13.4
600138
RP13 cromosoma
17p13.3
600059
RP17 cromosoma
17q22-q24
600852
Autosomica Recessiva (ar)
RP12 cromosoma
1q31-q32.1
600105
RP14 cromosoma
6p21.3 RP14;
600132 GUCA1(?)
PDEB cromosoma
4p16.3 PDEB;
180072 b-PDE
CNCG cromosoma
4p14-q13 CNCG;
123825 g-CNCG
PDEA cromosoma 5q31.2-q34 PDEA;
180071 a-PDE
RP16 cromosoma 14
Legata al cromosoma X (Xar)
RP2 cromosoma
Xp11.3
312600
RP3 cromosoma
Xp21.1
RPGR (retinitis pigmentosa GTPase regulator)
RP6 cromosoma Xp21.3-p21.2
RP15 cromosoma Xp22.13-p22.11
Digenica
ROM1 cromosoma
11q13 ROM1;
180721 ROM1
________________________________________________________________________________
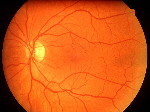 La Retina
La Retina
I CONI, così chiamati per la loro forma, che recepiscono soprattutto i particolari delle immagini ed i vari colori.
I BASTONCELLI, dalla linea allungata ed affusolata, i quali reagiscono prevalentemente al contrasto fra il chiaro e lo scuro ed al movimento degli oggetti.
La parte centrale della nostra retina (denominata macula) presenta moltissimi coni e pochi bastoncelli, essa ci permette, ad esempio, di leggere o di ammirare le sfumature di un quadro. Nella zone periferica sono invece assai più numerosi i bastoncelli, grazie a questi possiamo avvertire un pericolo con la coda dell'occhio, per esempio il movimento di una macchina che sta per investirci, anche se non siamo in grado di distinguere i particolari dell'automobile.
I Sintomi
I principali sintomi che possono indurre il medico a sospettare di trovarsi di fronte ad un caso di Retinite Pigmentosa sono essenzialmente due:
Cecità Crepuscolare e
Notturna
![]()
Cioè la difficoltà a vedere in condizioni di scarsa illuminazione (muoversi e guidare di sera o di notte) o problemi di adattamento nel passare dagli ambienti illuminati a quelli oscuri (entrare in una sala cinematografica buia). Questo fenomeno è dovuto al fatto che, almeno per la maggior parte dei casi, la malattia nelle prime fasi dello sviluppo aggredisce prevalentemente i bastoncelli.
Restringimento del Campo
Visivo ![]()
Si manifesta con la difficoltà nel percepire gli oggetti posti lateralmente, oppure con l'inciampare nei gradini o negli ostacoli bassi. Per farvi un'idea del disagio a cui il malato va incontro, potete immaginare di vedere costantemente il mondo da uno spioncino o dal buco della serratura. L'alterazione del campo visivo è progressiva e può giungere ad interessare anche la parte centrale dell'occhio, con perdita del visus. La velocità di progressione della malattia e l'età di comparsa dei sintomi variano in relazione a molti fattori tra cui il modello di trasmissione genetico.
Aumentata sensibilità
all'abbagliamento
![]()
Le degenerazioni retiniche provocano spesso un'elevata sensibilità all'abbagliamento. I contrasti svaniscono e diventa difficile percepire l'ambiente circostante.
La Diagnosi
La diagnosi di RP in presenza di tutti i sintomi classici è
facile ed è di pertinenza dell’oftalmologo. Per
diagnosticare la malattia vengono generalmente effettuati l'esame del fondo
dell'occhio e la sua fotografia, l'esame del campo visivo,
l'elettroretinogramma, la fluorangiografia, l'esame del visus.Può essere diagnosticata fin dall’infanzia, nell’adolescenza e, non di
rado, anche in età adulta. Nei casi dubbi, la diagnosi si basa su tutti i dati
clinici ottenibili (età di esordio, modalità evolutive, eventuale associazione
con altri sintomi oculari od a carico di altri organi ed apparati), e su di un
approfondito studio elettrofisiologico (elettroretinogramma ed
elettroculogramma) ed adattometrico. Sono esami utili e complementari lo studio
del senso cromatico e la fluoroangiografia retinica. E’ necessario inoltre
esaminare tutto il nucleo familiare, all scopo di definire il tipo di
trasmissione ereditaria.
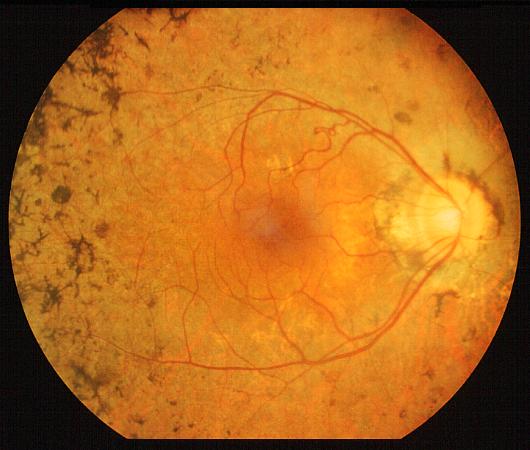
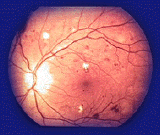
L'esame del Fondo dell'Occhio
Ha lo scopo di valutare l'apparenza morfologica della retina e di ricercare la presenza di caratteristiche macchie di pigmento sulla superficie retinica, che nella malattia hanno un aspetto caratteristico detto ad osteoblasti. Talune forme di Retinite, pur presentando gli stessi sintomi, non sono però caratterizzate dalla presenza di macchie sul fondo dell'occhio.
L'esame del Campo Visivo
Permette di valutare la sensibilità retinica ad uno stimolo luminoso nelle varie zone della retina stessa. È utile per avere una documentazione oggettiva delle difficoltà percepite dal paziente.
L'Elettroretinogramma
Consiste nella registrazione dell'attività elettrica della retina in risposta a particolari stimoli luminosi, permettendo di valutare in modo distinto la funzionalità dei due diversi tipi di fotorecettori (i coni ed i bastoncelli). L'elettroretinogramma è un esame molto importante per diagnosticare la Retinite Pigmentosa, poichè anche quando la malattia è ancora nella fase iniziale, il tracciato che ne deriva è quasi sempre estinto e molto appiattito.
La Fluorangiografia
Si attua con l'iniezione per via endovenosa di una sostanza fluorescente e nella successsiva fotografia della retina in tempi diversi. Infatti tramite la circolazione sanguigna, la sostanza fluorescente giunge sino alla retina, rendendo visibili, colorandole, le arterie, i capillari e le vene, nonchè lo stato funzionale delle loro pareti.
L' Esame del Visus
Permette di valutare l'acutezza visiva nella porzione centrale della retina. Consiste nella lettura di caratteri di varia grandezza alla distanza di 5 metri.
Il Decorso
Il decorso della malattia ha una durata estremamente variabile ma è comunque sempre progressivo ed invalidante. Nella maggioranza dei casi i sintomi precedentemente descritti si aggravano, il campo visivo si restringe sempre di più fino a chiudersi completamente. Compaiono poi altri disturbi come l'abbagliamento, l'incapacità di distinguere i colori, ed una particolare forma di cataratta. L'esito finale è purtroppo in molti casi la cecità assoluta.
Terapia
Sono stati proposti fino ad oggi una miriade di protocolli
terapeutici aventi un effetto sul metabolismo del fotorecettore (Vit.A,
antocianosidi, lenti speciali ecc.), ma nessuno si è dimostrato in grado nè di
guarire nè di modificare la progressione della malattia. Secondo
alcune ricerche, la terapia con ossigeno potrebbe essere di notevole importanza
per i dati sperimentali che confermano come l'ossigeno influisce positivamente
riducendo il tasso di morte cellulare dei fotorecettori. Tutti questi studi sia
effettuati con cellule isolate che con animali da esperimento hanno evidenziato
l'efficacia dell'ossigeno nel salvataggio dei fotorecettori e di converso la
tossicità per I fotorecettori di ambienti poveri d'ossigeno anche per retine
normali.
E' attualmente in corso di stampa un lavoro a livello internazionale
sull'utilizzo dell'OTI nella retinite pigmentosa con un follow up di tre anni
nel quale senza ombra di dubbio l'OTI determina un incremento dell'ampiezza
dell'elettroretinogramma statisticamente significativo rispetto al gruppo di
controllo del 100%, anche se ciò non è immediatamente riferibile ad un
miglioramento delle caratteristiche funzionali. Inoltre dopo tre anni di terapia
nessuno dei pazienti era peggiorato, mentre nel gruppo di controllo il 62% dei
pazienti presentava un peggioramento. Il danno dei fotorecettori nella Retinite
pigmentosa ha diverse origini, la principale coinvolge la fototrasduzione che
comporta differenti passi mediati da più proteine che se difettose geneticamente
possono portare a morte prematura il fotorecettore stesso per un'alterazione
della funzione. Molte forme di Retinite pigmentosa sono associate con mutazioni
puntiformi o con microdelezioni di geni così come altri deficit genetici che
hanno in comune un'alterazione del metabolismo del fotorecettore. Inoltre sono
state identificate mutazioni che causano la retiniate pigmentosa che alterano
proteine interferiscono direttamente con i meccanismi ossidativi sia
direttamente come la Rodopsina che indirettamente attraverso la fosfodiesterasi
la periferina o la proteina di membrana dei bastoncelli. La diversa causa
genetica comporta una certa eterogeneità clinica, questa è caratterizzata da una
riduzione del letto capillare che provoca un ulteriore riduzione della pressione
parziale di ossigeno a livello della coroide e della retina. Ciò è
particolarmente evidente nelle regioni immediatamente adiacenti ai fotorecettori
che si presentano ricchissime di mitocondri che ricordiamo sono gli organelli
deputati alla respirazione cellulare.
Studi sperimentali hanno
dimostrato che i fotorecettori retinici, in presenza di rodopsina anomala o di
alterazioni della fototrasduzione, sono molto sensibili alla disponibilità di
ossigeno, che viene richiesto in maggiore quantità per garantire una funzione
visiva sufficiente. Studi in vivo effettuati da Stone su topi hanno dimostrato
che soprattutto in una fase critica cioè nelle prime fasi di sviluppo l'ossigeno
è in grado di ridurre parzialmente il tasso di apoptosi (morte cellulare
programmata), mentre una esposizione a bassi livelli di ossigeno è in grado di
provocare la morte dei fotorecettori nella retina di ratti normali. In
precedenza in uno studio pubblicato nel 1989 abbiamo riscontrato che uno stato
di ipossia cronica come nelle broncopatie provoca un progressivo danno retinico
facilmente individuabile con l'elettroretinogramma che svela una precoce
riduzione in ampiezza. Studi di Stone ed altri autori hanno inoltre riportato
che condizioni di iperossia transiente possono causare un incremento dell'ERG
che dura parecchi mesi, ed in questo caso l'incremento di ossigeno dato dalla
camera iperbarica può completare le richieste metaboliche retiniche.
L'uso
dell'OTI rispetto alle condizioni di somministrazione di ossigeno a pressione
atmosferica è necessario in quanto in condizioni di respirazione di aria per
l'effetto Bohr dell'emoglobina la saturazione di ossigeno è al 97% mentre solo
una piccola parte di ossigeno viene trasportata attraverso il plasma in
relazione alla pressione parziale dell'ossigeno a livello del polmone. In
condizioni iperbariche la quantità di ossigeno disciolta nei fluidi incrementa
del 60%. Per poter spiegare l'attività dell'ossigeno iperbarico aumenta le
disponibilità metaboliche in differenti punti della catena della
fototrasduzione, prevalentemente a livello dell'inattivazione della rodopsina,
dell'attivazione della trasducina, sull'attività della guanilato ciclasi ed
infine in una migliore efficienza dei cambiamenti di conformazione della
rodopsina. Infine in un nostro studio in corso di realizzazione su 44 pazienti
affetti da RP trattati per un periodo compreso tra 5 e 10 anni dimostra che
pazienti in trattamento hanno solo il 20% di possibilità di perdere le
performances visive mentre pazienti non trattati presentano nel 60% diminuzioni
visive superiori al 30% differenza statisticamente significativa tra I due
gruppi.
A mio avviso esistono in letteratura studi clinici e sperimentali che
dimostrano l'efficacia dell'ossigenoterapia iperbarica nel ridurre la morte
cellulare e quindi nel migliorare la qualità della visione di pazienti che sono
ancora all'inizio della loro vita trattandosi di soggetti molto piccoli. In
questo caso la possibilità di continuare con migliori performances visive
garantisce un corretto sviluppo psicofisico ed una adeguata formazione
scolastica e professionale. Considerando l'importanza che ha la Retinite
pigmentosa che è stata inserita fra le cosiddette malattie sociali riteniamo che
sia necessario garantire la possibilità di usufruire attraverso il SSN della
terapia iperbarica almeno nell'ambito dei Policlinici Universitari e per
pazienti in età critica nei quali la terapia con l'ossigeno si è dimostrata una
valida risorsa contro questa malattia. Si ritiene in tal modo di ridurre
cospicuamente la rapidità di progressione della malattia e quindi ritardare la
soglia di riconoscimento dell' invalidità configurando un risparmio per lo
Stato.
Le prospettive della ricerca
Sebbene l'identificazione e la classificazione della Retinite Pigmentosa risalgano alla metà del secolo scorso, tuttavia ben pochi progressi concreti sono stati compiuti fino ad oggi, sia sul fronte delle cure possibili, sia su quello, altrettanto importante, della comprensione delle cause che la determinano e che ne regolano il decorso. Attualmente i filoni più promettenti della ricerca internazionale sono i seguenti:
La Genetica
Si propone di identificare il gene od i geni responsabili della malattia, per poter eventualmente intervenire in seguito con le moderne tecniche dell'ingegneria genetica.
I Trapianti
L'intento è quello di mettere a punto una tecnica che renda possibile il trapianto di tessuto retinico, o per lo meno l'innesto di cellule sane su retine malate.
L'Immunologia
Si prefigge di verificare alcune teorie che ipotizzerebbero un'alterazione del sistema immunologico alla basa della malattia.
La Sindrome di Usher
È una forma ancora più grave di Retinite Pigmentosa. In essa la malattia si presenta associata ad un sordomutismo presente fin dal momento della nascita. Come si può facilmente comprendere, questa patologia è particolarmente invalidante in quanto colpisce, oltre al senso della vista, anche quello dell'udito. Fortunatamente la Sindrome di Usher non è molto diffusa. Si pensa che coinvolga il dieci per cento di tutti i casi di Retinite Pigmentosa. Esistono infine anche altre sindromi che associano la Retinite ad altre menomazioni particolarmente gravi come ritardo mentale, polidattilia, microcefalia ecc. Si tratta comunque di affezioni, per fortuna, molto rare.
Esempi pratici dei disturbi visivi avvertiti dalle persone affette da R.P.
Sensibilità all'abbagliamento
![]()
Le degenerazioni retiniche provocano spesso un'elevata sensibilità all'abbagliamento. I contrasti svaniscono e diventa difficile percepire l'ambiente circostante. L'immagine mostra una strada, macchine chiare, persone e sullo sfondo una scalinata. Se gli occhi sono molto sensibili all'abbagliamento, il tutto si confonde in una superficie lattea e appiattita.
 |
 |
| Vista normale | Sensibilità all'abbagliamento |
In casi gravi nasce la sensazione di dirigersi verso un muro bianco. Ausili utili contro l'abbagliamento sono lenti filtranti speciali, protezioni laterali e un copricapo a tesa che ombreggi il viso.
Limitazione periferica del campo visivo
![]()
Le persone con limitazioni periferiche del campo visivo percepiscono in modo nitido e preciso ciò che fissano con lo sguardo. Nonostante la "vista a tunnel", spesso nelle zone marginali del campo visivo permangono piccole isole funzionali, le cui cellule visive forniscono qualche informazione supplementare per orientarsi. È caratteristico delle limitazioni del campo visivo periferico di non accorgersi di non notare ciò che non si vede perché laddove la retina non trasmette informazioni non è né chiaro né scuro.
Per capire questo fenomeno possiamo provare a percepire con gli occhi lo spazio dietro la nostra schiena.
Forse sappiamo cosa vi si trovi ma visivamente si tratta d'uno spazio impercepibile. Analogamente, davanti agli occhi di persone con limitazioni periferiche o sparse del campo visivo non sbucano macchie grige o nere per avvisarle di ciò che non hanno visto.
Una persona cerca di orientarsi. Guarda verso il punto in cui il treno si ferma e fissa le porte.
 |
 |
| Vista a tunnel | Vista normale |
Poi guarda il tabellone delle partenze per verificare se si tratti proprio del treno giusto.
Punta nuovamente lo sguardo sulla porta del treno appena scoperta. Questa sembra sparita poiché‚ viene nascosta da passanti che prima non aveva visto venire.
 |
 |
| Vista a tunnel | Vista normale |
Alla periferia del campo visivo i bastoncelli sono più numerosi dei coni. I bastoncelli permettono di vedere in condizioni di debole illuminazione o di luce scarsa. Per questo motivo, spesso le persone con limitazioni del campo visivo periferico soffrono di cecità notturna.

Vista normale (a sinistra e a destra) e cecità notturna (in
mezzo)
Cecità notturna non significa soltanto di non vederci bene di notte o con poca luce bensì anche di avere bisogno di molto tempo perché l'occhio si adatti al passaggio dal chiaro all'oscuro o ai cambiamenti d'intensità della luce. Anche la capacità di vedere i contrasti è molto diminuita.
 |
 |
| Vista normale | Adattamento lento |
ASSOCIAZIONI PER LA RETINITE PIGMENTOSA IN ITALIA
Retina Italia onlus è la Federazione nata per la tutela dei malati affetti da patologie distrofiche retiniche ereditarie, con particolare attenzione alla Retinite Pigmentosa. Alla Federazione aderiscono 11 Associazioni regionali e 2 Associazioni tematiche nazionali. La federazione è membro effettivo di RETINA INTERNATIONAL, fa parte, inoltre, della Consulta delle associazioni nazionali per lo studio delle malattie rare insediata presso il Ministero della Sanità.
Soci ed Associazioni Federate:
A.I.R.P.I. Onlus - Presidente: Maria Luisa Gargiulo
Casella Postale 4135 - Roma
Tel. 388.9321039
E-mail: airpi@tiscali.it
ATRI TOSCANA Onlus - Presidente: Simone Vannini
Via Del Paradiso, 55 - 50013 Campi Bisenzio (FI)
Tel. 055.89.51.998
E-mail: info@atritoscana.it
Sito: www.atritoscana.it
ARIS SICILIA - Presidente: Rocco Di Lorenzo
Via Amm. Gravina, 53 - 90139 Palermo
Tel. e fax 091.66.22.375
E-mail: aris@ipovisione.org
Sito web: http://www.ipovisione.org
RP LIGURIA Onlus - Presidente: Claudio Pisotti
Via Asiago, 3/R - 16137 Genova
Tel. 010.54.11.20
E-mail: rpliguria@libero.it - Sito web:
http://www.rpliguria.org
RETINA CALABRIA - Presidente: Carmela Petrelli
Via Roma - p. S. Ippolito 173
89013 Gioia Tauro (RC) Tel. 0966.51.174
E-mail: retinacalabria@email.it
RETINA CAMPANIA onlus - Presidente: Marco Colato
Via Pansini,5 - Napoli
Tel. 388 3648570
E-mail : info@retinacampania.it
http://www.retinacampania.it
R.P. TRIVENETO Onlus - Presidente: Rabito Roberto
Via Verdi 7 - 36030 Lugo di Vicenza (VI)
Tel. 0445.325164 – Fax 0445.395940
E-mail: rabitoroberto@libero.it
R.P. EMILIA ROMAGNA Onlus - Presidente: Mirella Bighi
Via Gandusio, 12 - 40127 Bologna
Tel. -Fax 051.24.67.05 - 051.63.90.645
E-mail: rpbologna@associazionerpbo.191.it
RP SARDEGNA Onlus - Presidente: Giuseppe Martini
Via Bruscu Onnis, 16 - Cagliari
Tel. 070.65.13.16
E-mail: rpsardegna@tiscalinet.it
APRP PUGLIA Onlus - Presidente: Gianfranco Taurino
Policlinico di Bari - Divisione Oculistica
Piazza Giulio Cesare,11 - 70124 Bari
Tel. 347.33.16.733 - E-mail : info@rppuglia.org
Sito web : http://www.rppuglia.org
RP ITALIA Onlus - Lombardia - Presidente: Elio Borgonovi
Piazza Quattro Novembre, 4 - 20124 Milano
Tel. 02.67.07.08.25 - Fax 02.67.07.08.24
E-mail: rpitalia@alice.it
ANS . ASS. SUBVEDENTI Onlus - Presidente: Carla Mondolfo
Via L.go Volontari del Sangue, 1 - 20100 Milano
Tel. - Fax 02.70.63.28.50
E-mail: info@subvedenti.it
Sito web: http://www.subvedenti.it
ADV Associazione Disabili Visivi - Presidente: Giulio Nardone
Via Lima, 22 - 00198 Roma
Tel. 06.85.50.260 - Fax 06.88.40.490
E-mail: presidenza@disabilivisivi.it
Sito web: http://www.disabilivisivi.it
Riferimenti
bibliografici
|
 |


Questa pagina è stata
visitata
volte dal 18.03.1999
(ultimo aggiornamento febbraio 2018)