


Le Neoplasie Maligne e Benigne
(ultimo aggiornamento febbraio 2018)
Tumori Oculari Benigni:
EMANGIOMA DELLA COROIDE
Il più
frequente tumore primitivo vascolare della coroide è l’emangioma cavernoso
circoscritto. Gli altri tumori vascolari primitivi della coroide
(l’emangiopercitoma e l’emangioma capillare) sono estremamente rari.
L’emangioma cavernoso coroideale può essere di tipo circoscritto o
diffuso. Quest’ultima forma può essere correlata alla Sindrome di
Sturge-Weber.
L’EMANGIOMA COROIDEALE
CIRCOSCRITTO
Definizione e classificazione
L’emangioma coroideale
circoscritto è un importante tumore vascolare che entra in diagnosi
differenziale con altre lesioni amelanotiche dell’uvea (melanoma amelanotico,
metastasi, osteoma, degenerazione maculare ecc.).
La forma circoscritta di questo tumore
benigno è raramente associata al nevus flammeus cutaneo che invece caratterizza
la sindrome di Sturge-Weber.
La
frequenza di questo tumore, di solito unilaterale, è di circa 30 volte inferiore
rispetto a quella del melanoma uveale anche perché molti casi, essendo
asintomatici, rimangono misconosciuti.
Caratteristiche cliniche e diagnostiche
Il tumore rimane generalmente misconosciuto
fino a quando diventa sintomatico. La comparsa di sintomi, calo dell’acuità
visiva e/o metamorfopsie, avviene generalmente nella terza o quarta decade di
vita.
L’esame obiettivo del segmento anteriore è sempre normale
così come la pressione intraoculare.
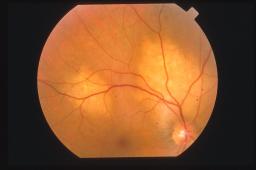
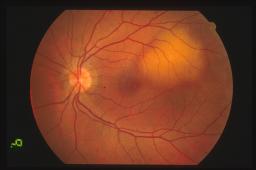
Oftalmoscopicamente il
tumore appare come una massa sottoretinica di un colore rosso arancio spesso
identico a quello della coroide circostante. Sulla superficie tumorale possiamo
ritrovare foci bianco-giallastri o più frequentemente accumuli di pigmento
sottoretinico a livello di epitelio pigmentato retinico.
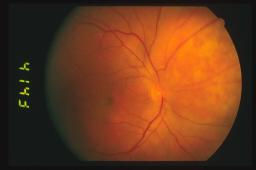
La
dimensione dell’emangioma coroideale varia circa da tre a 18 mm di diametro
massimo e da uno a 7 mm di spessore.
La
sede più frequente di riscontro dell’emangioma coroideale è la coroide
posteriore. In particolare l’80% ha sede al polo posteriore ed il rimanente 20%
è in genere parapapillare.
La presenza di un emangioma in sede
sottomaculare determina già nei primi anni di vita un'ipermetropia monolaterale
con rischio d'ambliopia. Queste caratteristiche devono quindi far sospettare,
nelle ambliopie anisometropiche dell’infanzia, la presenza di un emangioma
corideale sottomaculare.
Se il tumore è invece paramaculare, si
potrà avere un calo dell’acuità visiva in età adulta dovuto ad un sollevamento
sieroso del neuroepitelio retinico coinvolgente la fovea. Nell’adulto, alla
presenza di distacchi sierosi del neuroepitelio al polo posteriore, deve essere
quindi esclusa la presenza di piccoli emangiomi coroideali paramaculari.
Quando il
distacco del neuroepitelio persiste per lungo tempo si potranno osservare
alterazioni atrofiche dell'epitelio pigmentato retinico (“sindrome
gravitazionale”), degenerazioni cistiche ingravescenti della retina e zone
ischemiche retiniche periferiche con neovasi retinici al loro margine. In
presenza di tumori di grosse dimensioni si possono avere distacchi retinici
secondari totali.
In genere l’emangioma coroideale ha delle caratteristiche
oftalmoscopiche tali da consentire una diagnosi agevole.
Esistono tuttavia delle lesioni d'aspetto
morfologico non caratteristico che rendono difficile la diagnosi differenziale
con altre lesioni amelanotiche coroideali (melanoma amelanotico, metastasi,
corioretinopatia sierosa centrale, sclerite posteriore, osteoma della coroide,
degenerazione maculare senile, ecc.). In questi casi, oltre alla valutazione
oftalmoscopica binoculare diretta o indiretta (Infatti è fondamentale la visione
stereoscopica del fondo oculare che permette di visualizzare la “rilevatezza”
del tumore sul piano retinico ed il suo colore.), è necessario eseguire alcuni
esami strumentali aggiuntivi.
Per una corretta
interpretazione delle immagini diagnostiche ricavabili da indagini strumentali
bisogna tenere sempre presente la sede coroideale del tumore e le sue
caratteristiche istologiche. Infatti, l’emangioma è costituito da un gomitolo di
vasi sanguigni con scarsissimo tessuto di sostegno perivascolare in cui vi è un
flusso ematico rapido e vorticoso di molto superiore alla coroide sana
circostante.
Gli esami strumentali in grado di dare immagini patognomoniche
d'emangioma coroideale sono l’ecografia Ad e B Scan e l’angiografia con verde
indocianina.
L’ecografia A-scan standardizzata mostra picchi acustici
intralesionali ad alta reflettività pari a circa l’80-90% del picco retinico di
apertura. Quest'alta reflettività interna, è patognomonica di emangioma
coroideale, ed è determinata dalle numerose interfacie acustiche che
costituiscono le pareti dei vasi dell’emangioma.
In B-scan,
l’emangioma mostra sempre una forma ovalare a volte associata all’immagine di un
piccolo distacco del neuroepitelio sovrastante il tumore o di un sollevamento
sieroso retinico più ampio perilesionale. L’immagine è ad alta reflettività
paragonabile a quella del grasso retrobulbare e superiore a quella della coroide
circostante.Il tumore non invade mai la membrana di Bruch e quindi, immagini a
forma di fungo, sono sempre patognomoniche di
melanoma coroideale. L’immagine ecografica di calcificazioni intralesionali è
talvolta visibile nel contesto di un emangioma coroideale. Tali lesioni sono
però facilmente distinguibili dall’aspetto ecografico d'iperiflettività assoluta
dell’osteoma coroideale.
Un altro esame fondamentale, nella diagnosi
differenziale tra emangioma e lesioni amelanotiche della coroide, è
l’angiografia con verde indocianina che permette di visualizzare la circolazione
della coroide e dei tumori coroideali non pigmentati.
Nell’emangioma coroideale, l’angiografia con verde indocianina
mostrerà un rapido riempimento del tumore (iperfluorescenza precoce), dovuto al
flusso ematico intenso, con un'iperfluorescenza massima dopo circa 30 sec.
Dopo tale periodo vi è un
altrettanto rapido svuotamento dei vasi tumorali dal colorante con
un'ipofluorescenza del tumore rispetto alla coroide circostante.
Infatti, la scarsità di tessuto di sostegno perivascolare
dell’emangioma, determina un ridotto accumulo di colorante rispetto alla coroide
sana circostante.
Invece nelle altre lesioni amelanotiche
tumorali o pseudotumorali l’elevata quantità di tessuto rispetto all’esigua
vascolarizzazione interna determinano nell’angiografia con verde indocianina un
lento riempimento iniziale (ipofluorescenza precoce) e un accumulo maggiore di
colorante in sede extravasale
nelle fasi tardive (iperfluorescenze tardive) dell’esame.
Altri esami strumentali
utili nella gestione dell’emangioma coroideale sono l’angiografia a fluorescenza
e la tomografia coerente a radiazioni ottiche (OCT).
Queste
indagini strumentali, pur non fornendo informazioni patognomoniche, consentono
una valutazione qualitativa e quantitativa delle alterzioni retiniche secondarie
alla presenza del tumore o agli esiti del suo trattamento.
L’angiografia a
fluorescenza permette una visualizzazione del distacco sieroso del
neuroepitelio, che determina il calo dell’acuità visiva, e la scoperta di una
lesione coroideale adiacente o sottostante.
Inoltre le alterazioni
atrofiche “gravitazionali” dell’epitelio pigmentato retinico sono ben visibili
all’angiografia e dimostrano che la lesione è datata.
L’OCT è
un’indagine strumentale, di recente introduzione, che ci consente un rapido e
non invasivo studio degli strati retinici in senso qualitativo e quantitativo.
L’esame consente di evidenziare e quantificare il sollevamento del neuroepitelio
maculare secondario all’emangioma coroideale. Tali parametri saranno quindi
monitorati nel tempo per valutare follow-up naturale o dopo trattamento.
Istopatologia
Alla
valutazione istologica il tumore appare come una massa di vasi sanguigni
congesti. Possono essere prevalentemente di tipo capillare, cavernoso o misti.
La forma cavernosa è la più frequenter ed è caratterizzata da ampi vasi congesti
separati da sottili setti connettivali. La forma capillare è rara ed è
costituita da fini capillari separati da connettivo. Le alterazioni della retina
soprastante il tumore sono caratterizzate da degenerazioni cistiche retiniche,
macrofagi contenenti melanina o lipofuscina, metaplasie dell’epitelio pigmentato
retinico.
Trattamento
L’emangioma della coroide è un tumore benigno e quindi il
trattamento non è finalizzato alla distruzione del tumore a qualsiasi costo ma,
invece, al mantenimento della migliore acuità visiva per il maggior tempo
possibile.
Se il paziente non ha sintomi o calo dell’acuità
visiva il trattamento non è necessario ed il paziente andrà controllato nel
tempo.
Se un sollevamento del neuroepitelio determina un calo
dell’acuità visiva il trattamento dovrà essere preso in considerazione.
I trattamenti sono essenzialmente di due tipi: la laser terapia e la
radioterapia.
La scelta della migliore terapia per un determinato
emangioma dipende dal suo spessore, dal diametro, dall’ampiezza del sollevamento
sieroso del neuroepitelio, dalla presenza di danni irreversibili retinici
maculari e dalla localizzazione del tumore.
Lo scopo del trattamento fotocoagulativo
laser è di creare, sopra la superficie tumorale, delle cicatrici
retino-coroideali tali da consentire creare un'adesione della retina
all’epitelio pigmentato sottostante allo scopo di ridurre l’essudazione con una
conseguente diminuzione o scomparsa del sollevamento sieroso del neuroepitelio
ed un miglioramento dell’acuità visiva.
Questo tipo d'approccio terapeutico è
indicato per emangiomi di piccole e medie dimensioni. La fotocoagulazione può
essere intensa con spot confluenti sulla superficie del tumore oppure con spot
intensi ma radi sulla superficie tumorale. Quest’ultima opzione è consigliabile
per le lesioni paramaculari.
L’angiografia a fluorescenza consente un utile monitoraggio degli esiti
del trattamento laser.
Un nuovo approccio terapeutico mediante
laser a diodo è la Termo Terapia Transpupillare (TTT). Questo trattamento,
attualmente utilizzato con successo nella distruzione di melanomi coroideali di
piccole dimensioni, è stato eseguito in via sperimentale anche nell’emangioma
coroideale.
Il trattamento
alternativo al laser è la radioterapia a basse dosi eseguibile con diversi
approcci, radioterapia esterna, placche episclerali, protoni e gamma knife.
Questo trattamento determina nel tempo una distruzione dell’emangioma con una
risoluzione del distacco retinico secondario.
Le indicazioni per
l’utilizzo della radioterapia riguardano le lesioni sottomaculari con
sollevamento retinico e gli emangiomi di grandi dimensioni.
La
prognosi quoad vitam è eccellente essendo l’emangioma un tumore benigno. La
prognosi visiva dipende dalla sede, dall’ampiezza della lesione e soprattutto
dalla presenza, dall’estensione e dalla durata del sollevamento del
neuroepitelio maculare.
EMANGIOMA COROIDEALE DIFFUSO
L’emangioma
coroideale diffuso è un tumore benigno vascolare occupante la coroide posteriore
che si continua con la coroide circostante con margini mal definiti. L’emangioma
coroideale diffuso è presente nel 50% dei pazienti con la sindrome di Sturge
Weber ma può essere presente, seppur raramente, da solo.
A
differenza della forma circoscritta, quest'emangioma è generalmente
diagnosticato nella prima decade di vita a causa dell’ambliopia ipermetropica
che determina o per la presenza dell’emangioma cutaneo.
La
pupilla dell’occhio affetto ha un riflesso più rosso del controlaterale sano
mentre vitreo e cristallino sono trasparenti. Oftalmoscopicamente si apprezza un
diffuso ispessimento coroideale rossastro che meglio si evidenzia all’esame
ecografico in B-Scan.
La gestione
dell’emangioma coroideale diffuso è difficile a causa delle dimensioni
dell’emangioma e si basa sulla fotocoagulazione, la radioterapia o la
chirurgia.
Inoltre molto frequentemente nell’occhio affetto da
emangioma è presente un glaucoma cronico ad angolo aperto.
La
prognosi visiva è scarsa e la prognosi quoad vitam appare condizionata solo se
l’emangioma oculo-cutaneo è associato ad un emangioma leptomeningeo diffuso
(Sindrome di Sturge Weber).
EMANGIOMA CAPILLARE
L’emangioma capillare della coroide è una
variante istopatologica dell’emangioma coroideale circoscritto. La lesione è
costituita, contrariamente ai grossi vasi dilatati coroideali dell’emangioma
circoscritto, da vasi di piccole dimensioni.
In
alcuni casi è associato ad emangiomi capillari cutanei
EMANGIOPERCITOMA
UVEALE
Si tratta di un tumore rarissimo di cui sono riportati pochi casi in letteratura. L’emangiopercitoma è costituito da una proliferazione di periciti vascolari. L’aspetto oftalmoscopico è quello di un tumore coroideale amelanotico. L’angiografia e l’ecografia oculare non danno immagini patognomoniche della lesione. Istologicamente si tratta di un agglomerato di canali vascolari separati da tessuto costituito da periciti.
L’emangioma capillare si può presentare come una lesione solitaria
retinica, o può far parte della sindrome di von Hippel-Lindau in cui sono
associate angiomatosi sistemiche.
Tuttavia in entrambi i casi il
tumore appare clinicamente e istologicamente identico.
Definizione e classificazione
di cellule endoteliali e canali vascolari. La lesione è diagnosticata
in genere tra i 10 ed i 30
anni quando la lesione, essudando, determina
un calo dell’acuità visiva. La lesione può essere multipla nel 30% dei
casi.
La bilateralità e la molteplicità delle lesioni
implicano una mutazione genetica e l’appartenenza alla sindrome di von Hippel
Lindau.
Caratteristiche cliniche e
diagnostiche
Nello stadio precoce, l’emangioma periferico è molto piccolo e
oftalmoscopicamente lo s'individua a fatica ma lo si può localizzare più
agevolmente seguendo i vasi afferenti ed efferenti dilatati. Quando il tumore
s'ingrandisce appare un nodulo rosso-rosa con i caratteristici vasi afferenti ed
efferenti dilatati che arrivano fino alla papilla ottica.
Se il
tumore è invece in sede epipapillare o iuxtapapillare può apparire come un
nodulo a margini netti senza i vasi dilatati.
L’emangioma
capillare retinico può essere diviso nella forma essudativa e
vitreoretinica.
La forma essudativa è
caratterizzata da un'essudazione intraretinica e sottoretinica che inizialmente
è perilesinale e
successivamente alla crescita del tumore aumenta fino a coinvolgere la macula.
Nei casi più avanzati l’essudazione può determinare anche un distacco retinico
essudativo totale simile a quello della malattia di Coats.
La
forma vitreoretinica è invece caratterizzata da emangiomi retinici con
minima reazione essudativa ma con una reazione vitreale intensa che può
determinare la formazione di bande vitreali e quindi trazione vitreo retinica.
Queste trazioni possono determinare distacchi di retina trazionali ed evolvere
in ptsi bulbare.
L’emangioma capillare retinico periferico ha
un aspetto oftalmoscopico caratteristico. In presenza di una vasta essudazione
retinica coinvolgente il polo posteriore il tumore entra in diagnosi
differenziale con la malattia di Coats da cui tuttavia si differenzia per la
presenza costante del tumore e dei vasi dilatati.
Altre lesioni con cui entra in diagnosi
differenziale l’emangioma capillare retinico sono l’emangioma racemoso retinico,
l’emangioma cavernoso retinico, i macroaneurimi retinici con essudazione
ecc.
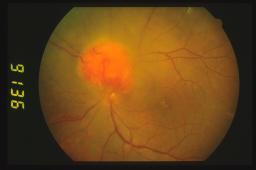
L’emangioma in sede peripapillare può entrare in diagnosi
differenziale con il papilledema e con la papillite. Un accurata anamnesi,
un’attenta oftalmoscopia binoculare ed un’angiografia a fluorescenza sono in
grado di differenziare le lesioni.
Tra gli esami strumentali la
fluorangiografia consente di valutare dinamicamente il riempimento e lo
svuotamento dei vasi afferenti ed efferenti e di rivelare la presenza di
altre lesioni di piccole dimensioni e misconosciute all’esame oftalmoscopico.
L’esame è inoltre una guida indispensabile al trattamento laser dell’emangioma
ed al suo follow-up.
L’ecografia oculare e l’angiografia con verde indocianina non
danno informazioni aggiuntive di particolare interesse clinico nella diagnostica
differenziale.
L’emangioma retinico consiste in una
proliferazione di capillari che sostituiscono a tutto spessore la retina. La
crescita può avvenire verso il vitreo (endofitico) o verso la coroide
(esofitico). La proliferazione benigna riguarda le cellule endoteliali ed i
periciti. All’interno del tumore può esistere una marcata proliferazione
fibrogliale.
Il
trattamento deve iniziare solo quando il tumore, mostrando segni di crescita,
aumenterà l’essudazione retinica. Se non cresce e non determina essudazione
retinica deve essere controllato oftalmoscopicamente ogni 3-4 mesi.
I trattamenti dipendono dalla sede, dimensioni del tumore, dalla
trasparenza dei mezzi diottrici e dal grado di fibrosi vitreoretinica.
Lo scopo del trattamento di questo tumore benigno, ma a crescita
lenta, è la sua distruzione per fermare l’essudazione intra e sottoretinica
maculare che è la causa del calo dell’acuità visiva.
Il
trattamento fotocoagulativo laser transpupillare viene eseguito prevalentemente
su tumori periferici di piccole e medie dimensioni. La tecnica utilizzata
consiste nell’utilizzare basse potenze e lunghi tempi di esposizione.
Il trattamento sarà diretto sul tumore
ed in un’unica sessione per gli emangiomi di piccole
dimensioni (< a un diametro
papillare).
Per gli
emangiomi di diametro maggiore più grandi si preferisce iniziare con un
trattamento perilesionale (doppia fila di spot) e
dopo trenta giorni circa si esegue il trattamento sul vaso afferente.
Successivamente si tratterà direttamente la massa tumorale fino alla sua
completa distruzione (fig.5).
Negli emangiomi periferici più
grossi, è consigliabile utilizzare la crioterapia con la tecnica del triplo
congelamento ed eventualmente ripetere il trattamento a distanza di tre
mesi.
Il successo del trattamento comporterà una scomparsa
degli essudati retinici ed anche
riduzione del calibro della
forma dei vasi retinici tumorali.
EMANGIOMA
CAVERNOSO RETINICO
Emangioma cavernoso retinico è un tumore vascolare benigno. Lo si
diagnostica più frequentemente nei giovani adulti come lesione solitaria oculare
o associata ad altre malformazioni cutanee ed intracraniche.
Oftalmoscopicamente appare come un grappolo di aneurismi di colore
rosso scuro generalmente nella retina periferica e più raramente vicino al disco
ottico.
A differenza dell’emangioma capillare retinico le
pareti vascolari non sono alterate e quindi viene a mancare la componente
essudativa perilesionale.
Istologicamente si tratta infatti
di vene retiniche dilatate e congeste.
Alla fluorangiografia
retinica i vasi rimangono ipofluorescenti nelle fasi precoci e solo nelle fasi
tardive gli aneurismi si riempiono di colorante.
Nella
maggior parte dei casi non è necessaria alcuna terapia ed il riscontro del
tumore vascolare è spesso casuale. In rari casi vi può essere un'emorragia
vitreale e tale complicanza può richiedere una terapia adeguata chirurgica e/o
laser.
EMANGIOMA RACEMOSO RETINICO
L’emangioma racemoso o cirsoide retinico è un’imponente
alterazione vascolare del circolo retinico
dovuta ad un’anastomosi diretta arteria-vena.
La valutazione oftalmoscopica mostra un caratteristico ammasso di
vasi tortuosi, congesti e dilatati a partenza dalla papilla ottica.
Alla fluorangiografia si dimostra la comunicazione arteria-vena
con un rapido riempimento del tumore ma senza accumulo di colorante o
essudazione.
L’evoluzione è molto lenta con complicanze
oculari rare (emorragie retiniche o occlusioni vascolari).
L’emangioma racemoso può essere una componente della sindrome di
Wyburn-Mason per cui il paziente
deve essere indagato per escludere la
presenza di queste alterazioni vascolari in altre parti del corpo (cervello,
orbita, mandibola ecc.).
TUMORE VASOPROLIFERATIVO DEL FONDO
OCULARE
Il tumore vasoproliferativo del fondo oculare (TVPFO) è una massa
rosso-rosa con vaso retinico afferente e venoso
efferente solo lievemente tortuosi ma non dilatati.
Clinicamente possiamo dividere il tumore in forma primaria e forma
secondaria.
La forma primitiva è in genere una lesione
solitaria, monolaterale, localizzata nel quadrante infero temporale.
La forma secondaria occorre in occhi con lesioni
predisponenti (uveiti, toxocariasi, retinite pigmentosa ecc.), bilaterale ed
associata ad una forte essudazione retinica.
La terapia
fotocoagulativa laser è riservata a lesioni piccole e con scarsa essudazione.
Per lesioni di grandi dimensioni o con intensa essudazione è preferibile la
crioterapia.
ASTROCITOMA RETINICO
L’amartoma astrocitico della retina è un tumore benigno composto
da cellule gliali in prevalenza astrociti.
Clinicamente esistono due forme. La forma solitaria retinica
non associata a sclerosi tuberosa e quella associata a lesioni amartomatose
multiple extraoculari (astrocitoma intracranico, angiofibroma cutaneo, chiazze
cutanee di depigmentazione, rabdomioma cardiaco, angiolipoma renale, ecc.)
tipiche della sclerosi tuberosa.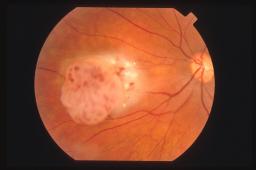
Oftalmoscopicamente la lesione appare in due forme principali. Una
sessile non calcifica ed una composta da sferule calcifiche che lo fanno entrare
in diagnosi differenziale con il retinoblastoma da cui differisce per la
mancanza di feeder vessels.
Fluorangiograficamente si
apprezzano i vasi tumorali ed un iperfluorescenza tardiva. Nei casi più atipici
può essere necessario la biopsia mediante ago aspirato.
TUMORI MIOGENICI
I tumori miogenici dell’uvea sono il rabdomiosarcoma ed il
leiomioma.
Il rabdomiosarcoma è un tumore maligno mesenchimale dell’infanzia. In letteratura esistono solo tre casi di rabdomiosarcoma dell’iride e dei corpi ciliari. Il tumore è indistinguibile alla valutazione clinico-strumentale da altre neoplasie.
Il leiomioma è un tumore benigno della muscolatura liscia colpisce in genere giovani donne. Clinicamente appare simile ad un melanoma amelanotico con vasi sentinella e con estensione sclerale. Il leiomioma è più frequentemente irideo e solo più raramente si possono trovare grossi tumori dei corpi ciliari a lenta crescita.
La terapia è chirurgica od osservazionale.
TUMORI NEUROGENICI
I tumori neurali che originano nel tratto uveale sono lo schwannoma, il neurofibroma.
Lo schwannoma (neurilemoma) è un tumore benigno delle guaine dei nervi ciliari del tratto uveale. Clinicamente appare come una neoformazione coroideale, prevalentemente non pigmentata, indistinguibile da un melanoma uveale.
Istologicamente è una proliferazione di cellule di Schwann e può presentarsi come forma solitaria o molto più raramente associata alla neurofibromatosi.
Il neurofibroma occorre in genere associato alla neurofibromatosi di von Recklinghausen. Si tratta di un amartoma di cellule gliali e melanocitiche a livello di iride (noduli di Lisch) e a livello di coroide.
Definizione e Classificazione
L’osteoma della coroide è un tumore benigno dell'uvea
costituito da tessuto osseo. Data la rarità diquesto tumore non esistono dati
epidemiologici consistenti sulla sua incidenza o prevalenza. Tuttavia dalla
valutazione della letteratura esistente l’osteoma appare più frequente nelle
donne giovani (20-30 anni) senza predilezione di razza. La lesione è unilaterale
nel 75-80% dei casi.
Caratteristiche Cliniche
e Diagnostica
Oftalmoscopicamente
l’osteoma appare come una lesione sottoretinica placoide giallo-arancio
contenente accumuli di pigmento marrone. La lesione può essere localizzata in
sede peripapillare o al solo polo posteriore. Le dimensioni variano da alcuni
millimetri di diametro fino ad occupare tutto il polo posteriore. La forma può
essere rotonda od ovoidale con margini ben definiti ma irregolari. Quando il
tumore è bilaterale le dimensioni sono spesso asimmetriche. Sulla superficie del
tumore sono talvolta visibili dei ciuffi di vasi che originano nella parte
profonda del tessuto osseo. In alcuni casi si possono sviluppare, nel contesto
del tumore, delle membrane neovascolari o dei sollevamenti sierosi del
neuroepitelio.
L’aspetto
oftalmoscopico è spesso patognomonico di osteoma coroideale. Tuttavia il tumore
entra in diagnosi differenziale con altre lesioni tumorali o pseudotumorali
della coroide (melanoma amelanotico, metastasi, emangioma, degenerazione
maculare ecc.).
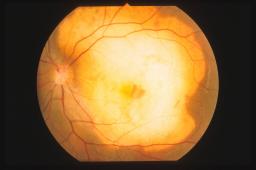
L’ecografia oculare B-scan è un esame strumentale
fondamentale nella diagnosi differenziale. Permette, infatti, di evidenziare
l’alta reflettività del tessuto osseo tumorale che blocca tutti gli ultrasuoni
determinando dietro alla lesione “ un’ombra acustica”. Riducendo gradualmente la
sensibilità dello strumento permane l’immagine iperriflettente del tessuto osseo
mentre si riduce quella del tessuto adiposo retrobulbare.
La fluorangiografia
retinica permette di visualizzare le alterazioni della coriocapillare e
dell’epitelio pigmentato retinico secondarie alla sostituzione della coroide da
parte del tessuto osseo tumorale. L'esame è quindi utile nella gestione delle
membrane neovascolari sottoretiniche e nei distacchi sierosi del neuroepitelio
retinico.
Molto più interessante è invece l’aspetto
dell’osteoma all’angiografia con verde indocianina. Quest'esame permette la
precisa visualizzazione dei margini coroideali della lesione e dei ciuffi di
vasi che dal profondo del tessuto osseo risalgono in superficie.
Istopatologia
L’osteoma è composto
di tessuto osseo maturo che sostituisce lo strato coroideale risparmiando parte
della coriocapillare. Nel tessuto tumorale sono presenti le trabecole ossee
contenenti ampi spazi cavernosi ricoperti da endotelio e piccoli vasi
capillari.
Le linee cellulari
presenti sono osteoblasti, osteociti e osteoclasti. Negli spazi intertrabeculari
midollari sono presenti elementi fibrovascolari, mastcellule e cellule
mesenchimali. La coriocapillare appare assottigliata e/o obliterata ed il
sovrastante epitelio pigmentato retinico appare assottigliato o atrofico
contenente accumuli di melanofagi contenenti pigmento.
La patogenesi di questo tumore è sconosciuta. Sono
state ipotizzate diverse origini (post-infiammatoria, post-traumatica o
coristoma) ma le caratteristiche clinicopatologiche della lesione (tessuto
osseo, età d'insorgenza e progressione) non confermano queste ipotesi.
Essendo un tumore
benigno a lentissima crescita, il trattamento consiste nell’osservazione
periodica e nel monitoraggio delle complicanze che possono ridurre l’acuità
visiva.
Un rapido deterioramento è generalmente legato
all’insorgenza di una membrana sottoretinica maculare. Questa è la complicanza
più temibile per la prognosi visiva e può essere trattata con fotocoagulazione
laser secondo criteri standard.
Un lento deterioramento
dell’acuità visiva è invece secondario ad un'alterazione progressiva dei
fotorecettori retinici dovuta all’alterazione della coriocapillare assottigliata
e occlusa dal tumore.
La prognosi visiva dipende quindi anche
dalla localizzazione del tumore. Non esistono invece differenze della prognosi
quoad vitam rispetto alla popolazione normale.
Tumori Maligni
MELANOMA OCULARE
I melanomi oculari (congiuntivali ed uveali) sono
tumori molto rari, caratterizzati da una prognosi spesso grave. I melanomi
uveali sono spesso asintomatici e
determinano disturbi aspecifici dell’acuità visiva solo quando raggiungono
dimensioni ragguardevoli o interessano l’area maculare. La miglior forma di
prevenzione e diagnosi precoce appare quindi il controllo annuale oftalmologico
del fondo oculare. Il coinvolgimento dell’occhio, organo di piccole dimensioni
ma con funzioni importantissime, impone poi la gestione del paziente presso
centri di alta specializzazione.
Definizione ed
incidenza.
I tumori maligni
congiuntivali sono rappresentati dai carcinomi a cellule squamose e sebaceo, dal
melanoma, dai tumori linfoidi, dal sarcoma di Kaposi. Tra questi, il melanoma,
che origina dai melanociti congiuntivali, è estremamente raro (meno del 2% di
tutti i tumori maligni oculari), con una incidenza compresa tra 0.024 e 0.052 nuovi casi ogni 100.000 abitanti,
pari a circa 1/40 di quella dei melanomi uveali. La patogenesi di questo tumore
è stata sempre fonte di controversie; attualmente si considera che esso origini
nel 75% dei casi da una melanosi acquisita primitiva (MAP), mentre nel restante 25% da un nevo
congiuntivale pre-esistente o de novo.
Diagnosi
La diagnosi differenziale
tra melanoma congiuntivale ed altre lesioni pigmentate benigne (nevo, melanosi,
melanocitosi) o pre-cancerose (melanosi acquisita primitiva) si basa
essenzialmente su storia clinica, morfologia della lesione e reperto
bioptico.
Nel nervo congiuntivale esistono spesso spazi cistici
intralesionali comunemente assenti nel melanoma. Inoltre l’incidenza del
melanoma è maggiore nella età di mezzo, mentre è raro nei primi anni di vita e
nell’adolescenza in cui è frequente il riscontro dei nevi. Il melanoma
pigmentato è anche facilmente differenziabile, mediante valutazione al
biomicroscopio, da quelle lesioni a sede sclerale (melanocitosi oculare o
oculodermica) che creano una pseudo pigmentazione
congiuntivale.
Tra le lesioni pigmentate della congiuntiva la più
importante è la melanosi acquisita primitiva (MAP). Si tratta di una lesione
piatta, quasi sempre monolaterale, che colpisce prevalentemente pazienti di
media età. Essa può aumentare di dimensioni o modificare la propria
pigmentazione nel tempo, ma, soprattutto, può trasformarsi in melanoma
congiuntivale; questo evento si verifica
50% dei casi di MAP che presentano atipie cellulari all’esame
istologico.
Nelle lesioni con un aspetto morfologico francamente
benigno l’approccio diagnostico si basa sulla valutazione clinica e sulla
documentazione fotografica, ripetute e confrontate nel tempo. Nelle lesioni
dubbie di piccole dimensioni è consigliata l’escissione chirurgica in toto e la
valutazione istologica. Nelle lesioni di maggiore dimensione è preferibile
eseguire biopsie multiple di aree con diversa morfologia. Le biopsie multiple di
lesioni maligne non peggiorano la prognosi. 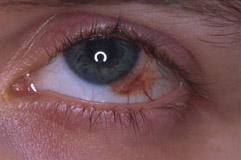
Il paziente affetto da melanoma congiuntivale
deve essere sottoposto, prima del trattamento locale, ad indagini diagnostiche
per escludere la presenza di metastasi. Queste possono interessare, per
diffusione linfatica, i linfonodi cervicali, sottomandibolari, preauricolari e
addominali, nonché il tessuto sottocutaneo periorbitario, e, per via ematica, il
fegato, lo scheletro, la parotide ed il sistema nervoso
centrale.
Robertson ha riferito su alcuni pazienti con metastasi
alle cavità nasali e paranasali; questa importante segnalazione suggerisce
l’esecuzione di una visita otorinolaringoiatrica.
Trattamento e risultati
L’approccio terapeutico è guidato dalle dimensioni e dalla
localizzazione del melanoma. Nei melanomi di piccole dimensioni localizzati in
sedi favorevoli (congiuntiva bulbare e perilimbare) è sufficiente una ampia
resezione chirurgica. Nei casi situati in sedi sfavorevoli (congiuntiva
palpebrale, fornice, caruncola e margine palpebrale) o di grandi dimensioni,
l’exenteratio orbitae, tecnica chirurgica radicale in cui si rimuove tutto il
contenuto orbitario con conseguente cecità e deturpazione del volto, era
considerata l’unico trattamento proponibile.
Un recente studio
retrospettivo ha tuttavia dimostrato che la sopravvivenza dei pazienti
sottoposti ad exenteratio orbitae non era migliore di quelli trattati con
exeresi chirurgica locale, suggerendo di limitare l’impiego della chirurgia
radicale ai soli casi che infiltrano l’orbita. Attualmente nei melanomi a
localizzazione sfavorevole si preferisce quindi eseguire un’asportazione
chirurgica totale della lesione con ampio margine di sicurezza ed associare la
crioterapia o la radioterapia.
Quest’ultima
utilizza isotopi beta emittenti, quali lo Stronzio-90 ed il Rutenio-106, che per
le loro caratteristiche energetiche, consentono di somministrare alte dosi a
livello della congiuntiva con risparmio del cristallino (meno del 5% della dose
totale). La mortalità a 5 anni
varia dal 14% al 27%; a 10 anni è del 30% circa. I fattori di rischio
statisticamente significativi sulla mortalità sono la localizzazione sfavorevole
(mortalità doppia rispetto alle altre localizzazioni), la presenza di cellule
miste, fusate ed epitelioidi (mortalità tripla rispetto alla sola cellularità
fusata), la multifocalità (mortalità quintupla rispetto alle forme monofocali).
Il grado di invasione profonda del tumore determina una mortalità più elevata,
soprattutto nel gruppo di pazienti con tumore in sede sfavorevole.
Il melanoma Uveale
Definizione ed incidenza.
Il melanoma uveale, che origina dai melanociti uveali
della cresta neurale, è il tumore maligno intraoculare più frequente dell’età
adulta; esso tende a crescere sia all'interno del bulbo, invadendo e
disorganizzando i tessuti intraoculari, sia all'esterno, infiltrando la sclera
ed i tessuti orbitari. Il melanoma uveale metastatizza a distanza unicamente per
via ematogena data l'assenza di vasi linfatici a livello
bulbare.
Nel 1979 Wilkes e coll. calcolarono una incidenza annuale
nella popolazione generale di 7 nuovi casi per milione di abitanti, con una
grande differenza in rapporto all’età: tre casi per milione al di sotto dei 50
anni, 21 casi al di sopra. Attualmente la sua incidenza annuale negli Stati
Uniti è stimata essere di circa 6 nuovi casi per un milione di abitanti. Sulla
base di questi dati possiamo presumere che in Italia si verifichino circa 350
nuovi casi ogni anno. La sede di insorgenza piu' frequente è la coroide (85%),
seguita dai corpi ciliari (10%) e dall'iride (5%).
Diagnosi
La diagnosi di melanoma coroideale è essenzialmente clinica. La diagnosi differenziale con le altre lesioni pigmentate uveali (nevo, melanocitoma, ipertrofia dell’epitelio pigmentato retinico, emorragia coroideale, neovascolarizzazione, eccetera) si basa sulla valutazione oftalmoscopica da parte di un oftalmologo esperto. Come esami accessori possono essere utilizzati l’angiografia a fluorescenza e/o con verde indocianina e l’ecografia oculare A/B scan.
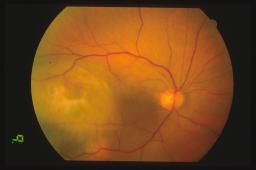
Le diagnosi differenziali più impegnative riguardano
i tumori amelanotici (nevi, emangiomi, metastasi, osteomi) e quei casi in cui
non è possibile visualizzare la lesione a causa dei mezzi diottrici non
trasparenti (lecomi corneali, cataratta, emovitreo, distacco di retina). In
tutti questi casi è raccomandabile l’uso di metodiche sofisticate quali
tomografia computerizzata, risonanza magnetica nucleare,
radioimmuno-scintigrafia o tomografia ad emissione di positroni. La storia
naturale della malattia, documentata in qualche caso dalla letteratura, è
invariabilmente caratterizzata dallo sviluppo di metastasi a
distanza.
Le sedi preferenziali sono il fegato (92% dei casi), il
polmone (31%), lo scheletro (23%), la cute (17%) ed il sistema nervoso centrale
(4%). Il tempo di comparsa dei secondarismi è estremamente variabile (da 2 mesi
a 30 anni); solitamente la loro comparsa porta al decesso entro un anno.
Prognosi
La classificazione citologica del melanoma uveale è un
importante fattore prognostico. Sono stati identificati tre tipi di cellule:
cellule fusate A, cellule fusate B, cellule epitelioidi. Sulla base di questa
suddivisione Callender classificò i melanomi in 6 gruppi, con prognosi
differente: a cellule fusate A e B, fascicolari, misti, necrotici ed a cellule
epitelioidi.
Più recentemente, McLean in un ampio studio
retrospettivo ha riclassificato su base prognostica i melanomi in tre gruppi:
nevi a cellule fusate, melanomi a cellule fusate e melanomi a cellule miste
(fusate ed epiteliodi). La mortalita' a 10 anni variava dallo 0% per i nevi a
cellule fusate a più del 50% per i melanomi a cellularità mista. Sono stati
individuati altri fattori in grado di influenzare la prognosi quoad vitam, oltre
alla citologia.
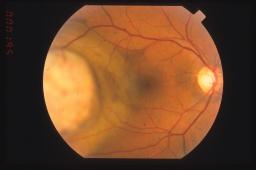
I piu' importanti sono: l'età del paziente, la localizzazione del tumore, il limite del margine tumorale anteriore, il diametro massimo tumorale, l'integrita' della membrana di Bruch e l'infiltrazione sclerale. A titolo puramente esemplificativo si può ritenere che un paziente anziano con un melanoma esteso ai corpi ciliari, con rottura della membrana di Bruch e con infiltrazione sclerale ha prognosi pessima.
Trattamento e risultati
L’evidenza anatomica di un tumore completamente
contenuto nel guscio sclerale e l'assenza di vasi linfatici bulbari hanno
giustificato per anni l'utilizzo dell'enucleazione quale unica metodica
terapeutica. Tuttavia, nonostante l'apparente radicalità dell’intervento e
l'assenza di metastasi al momento del trattamento, l’analisi di ampie casistiche
retrospettive evidenziava un elevato tasso di mortalità: il 35% a 5 anni, il
57% a 10 anni ed il 60% a 25
anni.
McLean e Zimmermann, in uno studio retrospettivo condotto su
3432 casi, evidenziarono la presenza di un picco di mortalità a circa due anni
dall'intervento di enucleazione, dovuto forse ad una disseminazione di cellule
neoplastiche durante l'intervento chirurgico. Sulla base di questa ipotesi si
cercò di ridurre le conseguenze della manipolazione chirurgica del bulbo oculare
sottoponendolo a congelamento durante l'intervento, o
ad irradiazione, con dose di 20 Gy, 24/48 ore prima
dell'enucleazione.
Questi accorgimenti non hanno tuttavia
dimostrato alcuna influenza sulla prognosi quoad vitam del paziente enucleato.
Vennero quindi sviluppate terapie conservative in alternativa all'enucleazione
(osservazione periodica di piccole lesioni, trattamento laser, resezione
chirurgica e radioterapia) in grado di garantire al paziente, a parità di
sopravvivenza, il mantenimento in sede del bulbo oculare con un eventuale
residuo visivo. L'osservazione periodica viene riservata alle lesioni di piccole
dimensioni (spessore inferiore a 3mm) ed è giustificata dalla constatazione che
alcune piccole neoformazioni precedentemente classificate come melanomi a
cellule fusate erano in realta' nevi a cellule fusate. Inoltre sono stati
documentati melanomi a crescita zero, definiti "dormant melanoma"
dagli Autori anglosassoni, che non crescono e non metastatizzano.
L'osservazione avviene mediante valutazione oftalmoscopica ma soprattutto
attraverso il confronto nel tempo di fotografie seriate della lesione. Il
trattamento fotocoagulativo laser viene attualmente utilizzato solo per lesioni
di piccole dimensioni (3mm di spessore massimo) a sede
periferica.
Le modalità di esecuzione variano
dalla fotocoagulazione transpupillare diretta con alte potenze, alla metodica
Low Energy-High Exposure introdotta allo scopo di aumentare la profondità della
necrosi. Il fascio di luce laser viene utilizzato per via transpupillare anche
nella terapia fotodinamica e nella termoterapia. La sopravvivenza dei pazienti
dopo questo tipo di trattamento appare sovrapponibile a quella ottenuta con le
metodiche più demolitive. 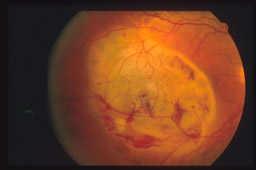
La resezione chirurgica locale del tumore,
introdotta da Foulds, comporta l'asportazione del tumore dall'esterno. E' una
tecnica chirurgica di difficile esecuzione e limitata ai melanomi dei corpi
ciliari e dell’iride. Il trattamento conservativo del melanoma uveale
attualmente più utilizzato è la radioterapia. Lo scopo della radioterapia è
quello di sterilizzare il tumore inibendo la capacità replicativa
cellulare.
Il melanoma uveale viene considerato radioresistente e
per il suo trattamento devono essere utilizzate dosi elevate di radiazioni
(50-60 Gy), in genere mal tollerate dalle strutture intraoculari più
radiosensibili (cristallino, nervo ottico, retina). Appare quindi indispensabile
utilizzare tecniche di irradiazione che consentano di somministrare alte dosi al
tumore risparmiando invece i tessuti peritumorali sani.
Già nel 1929 Moore tentò questo
approccio utilizzando una tecnica di brachiterapia interstiziale con aghi di
Radon impiantati direttamente nella massa neoplastica. Da allora ad oggi nuove e
più sofisticate metodiche sono state introdotte: la brachiterapia con placca
episclerale, la radioterapia con adroni (protoni e ioni elio) e la
radiochirurgia con Gamma Knife. La placca episclerale è costituita da un guscio
metallico di forma e dimensioni adeguate, contenente un isotopo radioattivo
(Cobalto 60, Rutenio 105, Iodio 125).
La placca viene suturata alla
sclera in corrispondenza del tumore permanendo il tempo necessario (sino a 10
giorni ) a somministrare una dose totale di almeno 100 Gy. Grazie al guscio
metallico l’irradiazione avviene prevalentemente verso il tumore con una
dispersione minima ai lati della placchetta. Mediante questa tecnica possono
essere trattate lesioni con spessore massimo di 5mm. Il controllo locale di
malattia è dell’85%, con la
conservazione di un buon grado di acuità visiva nella quasi totalità dei
pazienti. La radioterapia con adroni utilizza
generalmente protoni con energia di almeno 75 MV prodotti da un
ciclotrone.
Le caratteristiche fisiche dei protoni (picco di Bragg)
consentono di ottenere un fascio molto collimato in grado di cedere tutta la
dose terapeutica sul bersaglio ad una profondità voluta. Presso il Massachussets General
Hospital-Harvard Cyclotron Laboratory di Boston, dal 1976 ad oggi, sono stati
trattati oltre 2200 pazienti. La dose totale somministrata è stata di 70 Gy
equivalenti. L’analisi più recente è stata condotta su 1006 casi ed è risultata
in un controllo locale a 5 anni del 96%; il 90% dei pazienti guariti ha
conservato l’occhio, il 50%
l’acuità visiva. La sopravvivenza a 5 anni è dell’80%.
Possono essere
trattati tumori di tutte le dimensioni ed in qualsiasi sede, ma i migliori
risultati sono stati ottenuti nelle lesioni con diametro inferiore a 16mm e
spessore inferiore ad 8 mm che interessavano la sola coroide. In qualche centro
l’adroterapia è stata praticata utilizzando ioni elio: è riportata una
percentuale di fallimenti locali assai bassa (2,4% a 5 anni ed oltre), nettamente inferiore
a quella ottenibile con altre forme di irradiazione localizzata.
La
Gamma Knife è una apparecchiatura complessa, che comprende, oltre l’unità
radiante, i sistemi per la localizzazione del tumore, la definizione del piano
di trattamento ed il controllo del trattamento. Il cuore della Gamma Knife è un
computer (Gamma Plan) direttamente interfacciato con i più sofisticati sistemi
di imaging neuroradiologici (tomografia computerizzata e risonanza magnetica ad
alta definizione). Sullo schermo del computer, al quale giungono direttamente le
immagini diagnostiche, l’operatore è quindi in grado di localizzare il tumore,
disegnarne i margini, individuare le parti anatomiche da non irradiare,
scegliere il collimatore di diametro adeguato alle dimensioni del tumore,
calcolare la dose di irradiazione (tempo per numero di shot) ed infine
visualizzare le isodosi del trattamento direttamente sull’immagine
neuroradiologica.

L'unità radiante contiene 201 sorgenti di cobalto-60 collocate in
un corpo emisferico centrale. Il raggio emesso da ciascuna sorgente di cobalto
viene accuratamente collimato e fatto convergere con precisione in un punto
comune, definito isocentro, corrispondente all'intersezione dei raggi nel centro
del casco collimatore. La distribuzione geometrica delle sorgenti e il sistema
di collimazione assicurano così dosi elevate all’isocentro, di cui possono
essere variate forma e dimensioni, con risparmio dei tessuti perilesionali
sani.
Le Mestastasi Oculari
Introduzione
Le metastasi uveali sono i tumori
intraoculari più frequenti, anche se la
loro presenza è spesso sottostimata.
Infatti, la valutazione autoptica di bulbi oculari di pazienti deceduti per
tumore ha evidenziato la presenza di metastasi coroideali, clinicamente non
rilevate in vita, nel 4% dei
casi.
L’uvea ha delle caratteristiche anatomiche peculiari. Infatti la sua
struttura vascolare è di tipo terminale a lobuli. Queste caratteristiche ne
fanno una specie di rete filtrante in cui eventuali cellule tumorali presenti
nel sangue, si impiantano e
sviluppano la metastasi.
Tumori Primitivi
Al momento del riscontro di un secondarismo uveale la storia
clinica di un pregresso tumore primitivo è presente nel 70% dei casi. Nel
restante 30% non vi è invece un’anamnesi positiva per patologie tumorali
pregresse.
In questi pazienti una successiva valutazione oncologica sistemica
rivela la presenza del tumore primitivo solo nel 50% dei casi.
Quindi nel 17%
dei pazienti in cui viene diagnosticata una metastasi uveale, non si riesce ad individuare la sede del
tumore primitivo da cui è originata.
I tumori che più frequentemente determinano
metastasi coroideali sono i carcinomi. I più frequenti sono il tumore della
mammella (47%), del polmone (21%), del tratto gastrointestinale (4%), del rene
(2%), della pelle (2%), della prostata (2%), altre sedi (4%) e sede sconosciuta
(17%).
Le donne rappresentano il 70% dei pazienti affetti da
secondarismi uveali in virtù dello spiccato tropismo per la coroide che ha il
carcinoma della mammella.
L’età media dei pazienti è tra i 40
ed i 70 anni e dipende dalle caratteristiche epidemiologiche del tumore di
origine.
Caratteristiche Cliniche
La
sede più frequente delle metastasi uveali è la coroide (90%), i corpi ciliari
sono interessati nel 20% e l’iride nel 10%. La lesione è infatti unica nel 70% dei casi mentre nel restante 30%
può essere multifocale.
La
metastasi uveale può presentarsi bilateralmente circa nel 30% dei casi.
La multifocalità e la bilateralità rappresentano due caratteristiche
patognomonica di secondarismo molto utili nella diagnosi differenziale.
La localizzazione sottomaculare è presente nel 12% dei casi, mentre
nell’80% la sede è tra macula ed equatore.
Solamente l’8% delle metastasi sono post equatoriali.
Oftalmoscopicamente la metastasi si presenta come una lesione
giallastra con una forma placoide. Lo spessore medio ecografico è di 3mm ed il
diametro medio è di circa 9mm.
Nelle lesioni più spesse vi è un
distacco sieroso della retina sovrastante la massa con alterazioni dell’epitelio
pigmentato retinico. Questo tipo di alterazione accompagna le metastasi nel 75%
dei casi.
Diagnosi differenziale
La
diagnosi di metastasi coroideale è solamente clinica e si basa principalmente su
un’anamnesi clinica positiva per tumore primitivo e sulle caratteristiche
morfologiche della lesione.
La
diagnosi differenziale di una metastasi è verso altre lesione amenalnotiche
uveali che in ordine di frequenza sono: il melanoma amelanotico, l’emangioma
coroideale, il nevo amelanotico coroideale, sclerite posteriore, osteoma
coroideale, infiammazioni corioretiniche granulomatose e più raramente
degenerazioni maculari essudative.
Gli esami oftalmologici
strumentali (ecografia oculare, fluorangiografia retinica, angiografia con verde
indocianina) possono fornire indicazioni tra loro complementari sull’aspetto
morfologico della lesione. Tali parametri tuttavia servono più a escludere certe
patologie più che a essere patognomoniche di metastasi.
Anche
l’assenza di un tumore primitivo alla diagnosi e addirittura dopo la valutazione
sistemica oncologica non esclude la natura metastatica della lesione uveale.
Trattamento
Se
il tumore era misconosciuto il trattamento iniziale riguarderà il tumore
primitivo e successivamente, in funzione della prognosi quoad vitam, la
metastasi oculare. Se il tumore è noto il trattamento sarà sistemico e
locale.
Infatti lo scopo della terapia è quello di sterilizzare
la metastasi coroideale, in modo che essa stessa non determini ulteriori
disseminazioni per via ematogena, e distruggere eventuali metastasi sub cliniche
sistemiche.
Questo obiettivo viene perseguito con un primo
approccio chemioterapico sistemico. Se non c’è risposta terapeutica adeguata
alla chemioterapia o la lesione è a sede sottomaculare si deve utilizzare la
radioterapia esterna (3000-4000 cGy) con dosi iperfrazionate allo scopo di
ridurre al minimo gli effetti collaterali comunque inevitabili nel tempo.
Prognosi
In
genere la prognosi per questi pazienti non è buona con una sopravvivenza media
di 18 mesi dalla diagnosi. Tuttavia le pazienti con carcinoma della mammella
presentano una prognosi quoad vitam migliore soprattutto se la metastasi
coroideale era solitaria e prontamente trattata.

Questa pagina è stata
visitata
volte dal 18.03.1999